Author: OneAngstrom
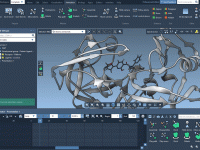
Mastering Bond Specification with Custom Attributes in SAMSON
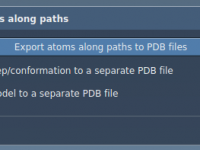
Streamlining Path-Based Atom Trajectory Exports in Molecular Modeling
Tailoring SAMSON: Customizing Your Molecular Modeling Workspace
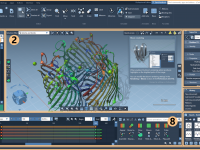
Simplifying Molecular Modeling Visuals: Master the ‘Shown’ Animation in SAMSON
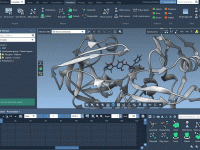
Enhance Your GROMACS Preparations with Custom Index Groups
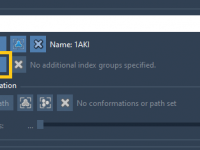
Mastering the Rock Animation in Molecular Modeling
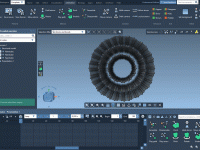
Streamline Molecular Modeling with the SAMSON AI /do Command
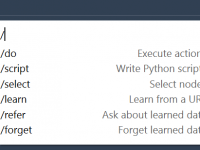
Efficient molecular modeling often involves repeatedly performing specific tasks, whether it’s selecting specific regions, modifying a visualization, or manipulating representations. These tasks, though essential, can slow down your workflow. This is where SAMSON AI’s /do command comes in, designed to…
Streamlining Molecular Visualizations with the Hide Animation in SAMSON
Molecular modeling and visualization often require precise control over the presentation of complex molecular structures. Imagine you need to isolate the behavior of specific molecular nodes without distractions, but manual adjustments for each keyframe make this task incredibly time-consuming. That’s…