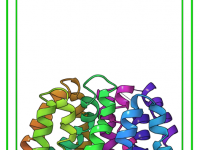
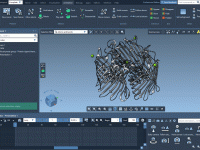
Simplifying Energy Minimization with SAMSON’s GROMACS Wizard.
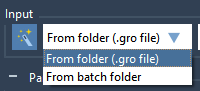
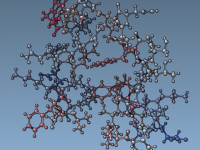
For molecular modelers, one major challenge is ensuring that molecular systems are geometrically stable before moving into simulations. This critical step—Energy Minimization—is essential to eliminate steric clashes and optimize the geometry of the system for further molecular dynamics simulations. With…