Bringing Molecular Models to Life with Interactive Simulations in SAMSON.
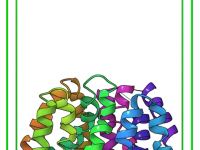
Molecular modeling experts often struggle with achieving realistic geometry adjustments in real-time, especially when working on complex systems. Wouldn’t it be rewarding to seamlessly manipulate your molecular structures while observing immediate feedback from physics-based simulations? With SAMSON, this is not…