Author: OneAngstrom
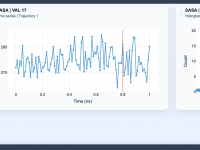
Tracking Solvent-Accessible Surface Area for Better Molecular Insights
Understanding Node Group Attributes in SAMSON
Streamlining Molecular Modeling with the Undock Animation
Molecular modeling involves intricate manipulation of molecular structures to simulate real-world behaviors. A frequent challenge molecular modelers encounter is repositioning groups of atoms or meshes effectively, especially when transitioning from docked (fixed) settings to exploring dynamic, spatial arrangements. Enter the…
Creating Constrained Simulations with the Simulate Animation in SAMSON.
Molecular modeling often requires controlling the positions of atoms in simulations to investigate specific conditions, functional behaviors, or engineered nanosystems. A common challenge many modelers face is performing constrained simulations effectively, ensuring the system behaves as desired. That’s where SAMSON’s…