Author: OneAngstrom
Refine Protein Conformations with the Ramachandran Plot in SAMSON
Effortless Molecule Visualization with 2D Depictions in SAMSON’s SMILES Manager
Unveiling Node Types in SAMSON: A Practical Guide
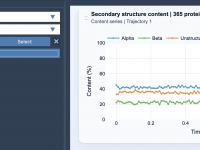
Understanding Protein Dynamics: Tracking Secondary Structure Content Over Time
Exploring File Format Compatibility for Molecular Modelers in SAMSON
In the world of molecular modeling, handling and exchanging data between different tools and platforms is often a tedious challenge. Whether it’s sharing molecular structures, trajectories, or simulation data, dealing with incompatible formats can complicate workflows and increase frustration. SAMSON,…