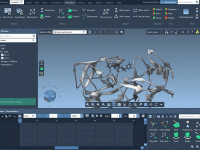
Author: OneAngstrom
Mastering Path Attributes in SAMSON’s Node Specification Language (NSL)
Protein Docking Starts with Proper Preparation: A Guide for SAMSON Users
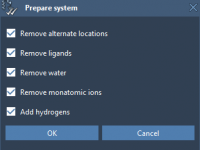
Avoiding Common Pitfalls When Setting Up Molecular Simulations in SAMSON
Mastering Constrained Simulations with SAMSON’s Simulate Animation
One of the key challenges molecular modelers frequently encounter is accurately simulating nanosystems while preserving specific constraints. Not all simulations are unconstrained—sometimes, you need precise control over atomic positions to reflect realistic conditions or achieve targeted designs. This is where…