





Category: Uncategorized
Visualizing the SARS-CoV-2 Spike’s Opening in 3D: A Practical Guide for Molecular Modelers
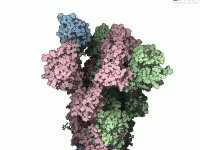
Understanding molecular motion is crucial in structural biology and molecular design, especially when studying protein-ligand interactions or conformational transitions. But modeling flexible structures like the SARS-CoV-2 spike protein can be technically challenging and time-consuming. Knowing how to simulate such motions…
Quickly Filter Visible Labels in Molecular Models Using SAMSON
Stop Wasting Clicks: Filter Render Presets in SAMSON with a Few Keystrokes
Need to track how atoms move? Here’s a way to record their path during animations.
Visualizing Molecular Motions Without Running Full Simulations
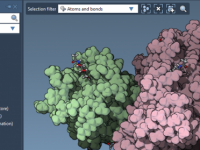
Computing conformational changes in large macromolecular systems is often time-consuming and computationally intensive. What if you need a representative trajectory between two known structures but can’t afford to run costly molecular dynamics simulations? For molecular modelers looking to visualize and…
A Clearer View: Exploring Structures with Progressive Clipping in SAMSON
Exporting Ligand Trajectories from Path Simulations Without the Clutter
Quickly Identify and Organize Labels in Your Molecular Models
Still Using the Wrong GROMACS Version?

If you’re working with GROMACS for molecular dynamics simulations, there’s a good chance you’ve encountered compatibility issues between versions. Whether you’re trying to reproduce published results, align with your lab’s computing environment, or troubleshoot strange errors, using a specific version…
Visualizing the SARS-CoV-2 Spike’s Opening in 3D: A Practical Guide for Molecular Modelers
Understanding molecular motion is crucial in structural biology and molecular design, especially when studying protein-ligand interactions or conformational transitions. But modeling flexible structures like the SARS-CoV-2 spike protein can be technically challenging and time-consuming. Knowing how to simulate such motions…
Quickly Filter Visible Labels in Molecular Models Using SAMSON
Stop Wasting Clicks: Filter Render Presets in SAMSON with a Few Keystrokes
Need to track how atoms move? Here’s a way to record their path during animations.
Visualizing Molecular Motions Without Running Full Simulations
Computing conformational changes in large macromolecular systems is often time-consuming and computationally intensive. What if you need a representative trajectory between two known structures but can’t afford to run costly molecular dynamics simulations? For molecular modelers looking to visualize and…
A Clearer View: Exploring Structures with Progressive Clipping in SAMSON
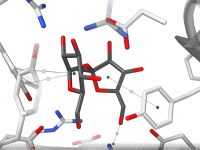
Exporting Ligand Trajectories from Path Simulations Without the Clutter
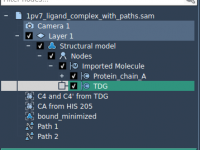
Quickly Identify and Organize Labels in Your Molecular Models
Still Using the Wrong GROMACS Version?
If you’re working with GROMACS for molecular dynamics simulations, there’s a good chance you’ve encountered compatibility issues between versions. Whether you’re trying to reproduce published results, align with your lab’s computing environment, or troubleshoot strange errors, using a specific version…