One of the key challenges faced by molecular modelers is effectively conveying spatial depth and structure in complex molecular scenes. Whether you’re presenting your work in a publication or simply trying to better understand a molecular structure, visual clarity can…
Collaborating on molecular modeling projects often means juggling email threads, cloud drives, file formats, and version mismatches. For molecular modelers, it can be frustrating to share large, complex structural data or simulation results in a streamlined and secure way—especially when…
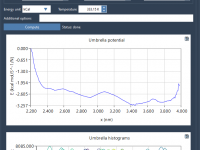
Running umbrella sampling simulations to determine the Potential of Mean Force (PMF) is a common and powerful approach in molecular modeling. But once trajectories are produced, a recurring challenge is understanding how well the reaction coordinate space is sampled. Poor…
Preparing a molecular animation that accurately conveys a binding mechanism or illustrates structural complementarity is often more work than expected. Whether you’re crafting a visual to accompany a publication or designing an animated explainer for your lab website, manually placing…
Working with large and complex molecular systems can often feel overwhelming, especially when you’re trying to isolate a specific part of your model—ligands, active sites, or particular side chains—for editing, analysis, or visualization. If you’ve ever found yourself switching endlessly…
When presenting molecular systems to colleagues or students, clarity matters. You want to walk them through a complex structure without clutter, and focus their attention on specific regions. But abruptly hiding parts of a structure can be visually jarring—especially in…
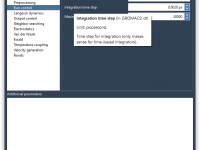
Anyone who uses GROMACS for molecular dynamics simulations knows that managing .mdp files can be time-consuming. These files control the behavior of your simulation, but editing them manually every time you switch between minimization, equilibration, and production phases is error-prone…
When working with protein conformational changes, it’s often necessary not only to generate realistic transitions between structures, but also to save and share these transitions for visualization, analysis, or subsequent simulation. If you’re using SAMSON’s ARAP Interpolator to create smooth…
Labels are a helpful way to annotate and organize structures in molecular modeling projects. But when your scene gets crowded with dozens of them, manual hiding or showing becomes inefficient—especially when you want to highlight just a specific subset. Fortunately,…
When working on large molecular modeling projects, it’s not unusual to find yourself navigating through dozens or even hundreds of files—simulation results, conformational variants, input parameters, and so on. Trying to locate a specific file becomes increasingly difficult as complexity…