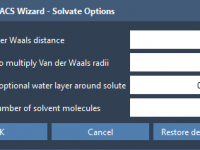
When setting up coarse-grained (CG) simulations in GROMACS, preparing the system correctly is essential—and solvent addition is a step that is often glossed over, or improperly configured. If you’re working with MARTINI-based CG models, especially those generated by Martinize2, there’s…
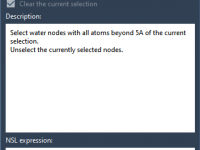
When preparing a biomolecular system for simulation, one common step is removing unnecessary water molecules. But not all waters are created equal. Active-site waters, often tightly bound and functionally important, should typically be preserved. So how can you remove only…
Molecular simulations often involve exploring the effect of different conformations of the same molecule. Whether you’re running umbrella sampling experiments or simply want to assess the stability of alternative poses, preparing the simulation for each conformation manually can be repetitive…
Capturing attention in molecular animations often means more than just rotating around a molecule. When presenting complex molecular scenes or creating scientific animations, you may want to create a controlled flythrough that smoothly zooms and shifts focus from one molecular…
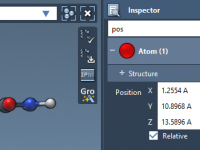
Molecular modelers working with complex structures often face a common challenge: how to find and edit only the relevant properties from a sea of attributes. Whether you’re tweaking atomic positions or checking element types, scanning through dozens (if not hundreds)…
When collaborating across institutions and disciplines, visibility and clarity of roles make a real difference. As a molecular modeler or computational scientist, you’ve probably shared files across cloud drives and sent endless emails introducing yourself and your expertise. But what…
When working with complex molecular systems in SAMSON, switching focus between different molecular components—like ligands, protein domains, or specific residue types—can become tedious. Typically, every time you want to reselect a region, you might need to dig through the Document…
When presenting molecular systems or creating visual walkthroughs in SAMSON, scientists and educators often run into a familiar challenge: how to create smooth, clear animations that allow viewers to follow complex structures horizontally across a scene. While rotating and zooming…
If you’ve ever worked with biomolecular structures, you’ve likely faced the challenge of identifying specific types of residues—such as acidic side chains, positively charged amino acids, or those with specific pKa ranges. These tasks are essential for setting up simulations,…
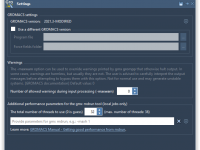
One common frustration among molecular modelers is the unresponsiveness of their computer when running local simulations. You’ve likely experienced it: you launch a simulation, and suddenly everything slows to a crawl. This often happens when computational tools like GROMACS run…