Streamline Molecular Modeling with Batch Protein Preparation in SAMSON
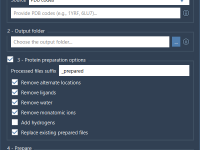
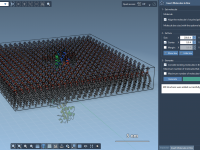
For molecular modelers handling large-scale datasets, preparing numerous protein structures manually can quickly become overwhelming. Resolving missing residues, adding hydrogens, or cleaning protein entries one by one is tedious and time-consuming. Thankfully, SAMSON offers a powerful solution: the Batch Protein…
A Quick Guide to Building Lipid Layers Around Proteins in SAMSON
Creating Enhanced Visual Representations in Molecular Modeling

Mastering the SAMSON Document View: A Closer Look
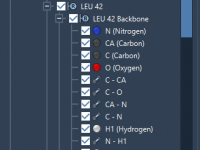
For molecular modelers, managing complex molecular hierarchies can be challenging. SAMSON’s Document View offers a structured, user-friendly way to handle intricate molecular data graphs. Whether you’re working on molecules, conformations, or Python scripts, this essential SAMSON feature simplifies navigation and…
Streamline Molecular Visuals with Smart Color Schemes in SAMSON
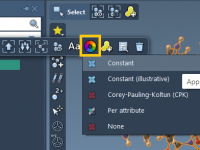
Working in molecular modeling often involves analyzing complex structures, where effective visualization is essential for understanding and communication. A common challenge is translating these intricate models into clear, visually meaningful representations. SAMSON, the integrative molecular design platform, offers a powerful…
Understanding Node Types in SAMSON: A Key to Molecular Modeling Organization
Unlock Flexibility in Docking: Setting Flexible Receptor Side Chains with SAMSON’s AutoDock Vina Extended.
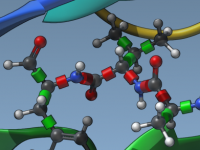
Docking studies are vital for molecular modeling, especially when exploring protein-ligand interactions. One persistent challenge is accounting for molecular flexibility—many docking protocols assume rigid receptor structures, which can overlook biologically relevant conformations. With AutoDock Vina Extended as part of the…
Efficiently Modeling Conformations with Path Attributes in SAMSON’s NSL
Molecular modeling often requires precise selection and manipulation of structural data, making flexibility and specificity critical for success. Whether you’re analyzing a pathway or focusing on specific structural conformations, SAMSON’s Node Specification Language (NSL) offers versatile tools to customize workflows.…