Mastering Molecular Bonds: Adjust Bond Orders in SAMSON.
One of the frequent challenges molecular modelers encounter involves managing and modifying bond orders within molecular structures. Whether you are modeling organic compounds, biomolecules, or new materials, ensuring precise bond representation is crucial for accurate simulations and analyses. In this…
Dive into the File Formats Supported by SAMSON
Discovering Protein Motions with Normal Modes Advanced in SAMSON
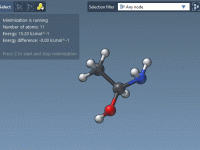
Tips for Using Measurement Labels Effectively in Molecular Modeling
In molecular modeling, accurate measurements are crucial for analyzing molecular structures, inspecting interactions, and performing structural adjustments efficiently. The Measure editor in SAMSON facilitates quick measurements of bond lengths, distances, angles, and dihedrals, while also allowing these measurements to be…
Maximizing Efficiency with Active Editors in SAMSON

A Practical Guide to Building Carbon Nanotube Models in SAMSON.

Carbon nanotubes (CNTs) have become a cornerstone in molecular modeling, nanotechnology, and material design. From nanosensors to molecular transport systems, their diverse applications make them essential for researchers. Yet, building customized CNT models efficiently can still be a challenge. With…
Simplify Molecular Labeling with ‘Label’ Attributes in SAMSON
Creating Smooth Transition Paths Between Protein Structures Using ARAP Interpolation
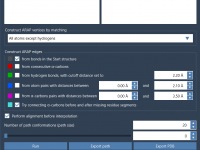
Protein modeling often requires understanding how a molecule transitions between two states. Whether you’re studying conformational changes, building realistic reaction coordinates for free energy simulations, or analyzing transition states, crafting a continuous, biologically meaningful path can be a daunting task.…