





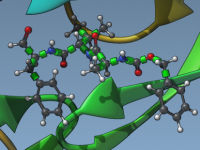
Why Validating Protein Structures Is Crucial for Successful Molecular Modeling
Simplifying NVT Equilibration with GROMACS Wizard in SAMSON
Easily Collaborate with Shared Molecular Documents on SAMSON Connect.

Simplify Molecular Modeling with Bond Attributes in NSL
Effortlessly Build Carbon Nanotube Models in SAMSON

Carbon nanotubes (CNTs) are a cornerstone of modern nanotechnology, with applications ranging from creating nanodevices and sensors to simulations of material properties or drug delivery systems. However, building accurate models of single-walled or multi-walled CNTs can be a time-consuming challenge…
Enhance Molecular Visuals with Depth of Field Effects in SAMSON.
Unlocking Molecular Motion: A Guide to Pathlines in SAMSON
Understanding Molecular Bond Types in SAMSON’s NSL
Easily Align Protein Sequences and Structures Using SAMSON’s Protein Aligner
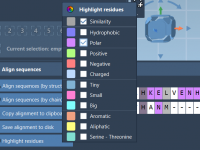
Aligning protein sequences and structures is critical for molecular modelers aiming to understand functional similarities, evolutionary relationships, or to design novel molecules. However, the process can be time-consuming, especially when working with multiple chains or complex models. Fortunately, SAMSON’s Protein…