Mastering Document Organization in SAMSON for Molecular Modeling
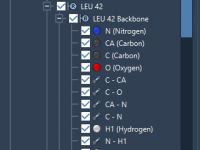
One persistent challenge for molecular modelers is managing and organizing complex structural data. Whether you’re working with molecules, scripts, conformations, or files, keeping track of this information in an efficient manner can significantly impact your productivity. SAMSON offers a solution:…
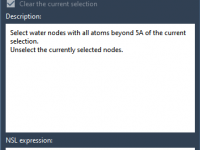
Understanding Segment Attributes in SAMSON’s Node Specification Language
Unlock Advanced Molecular Modeling with SAMSON AI’s Python Script Generator
Simplifying Cloud Job Management with SAMSON’s Job Manager
Mastering Path Recording for Molecular Animations
Enhance Your Modeling with SAMSON’s Dark Mode
Molecular modelers often spend extended periods analyzing molecular structures, tweaking settings, and running calculations. While the default interface settings in software platforms are often functional, they may not always provide the most comfortable experience—especially during long work sessions or in…