





How to Launch SAMSON on Your Operating System
Bringing Precision to Your Molecular Presentations with Pause Animations
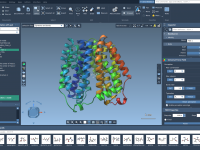
Simplifying Molecular Visualization with SAMSON’s Visual Presets
Streamline Molecular Visualization with Visual Presets in SAMSON
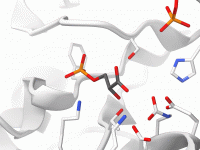
Are you a molecular modeler struggling to efficiently visualize complex molecular systems for your scientific research or presentations? The myriad of representations, colors, and detailed adjustments can make visualization overwhelming. With SAMSON, the process becomes significantly easier through the use…
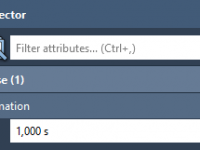
Understanding Property Model Attributes in SAMSON
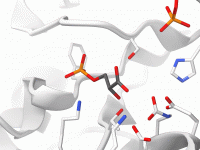
Generate Smooth Transition Paths Between Protein Conformations Using ARAP
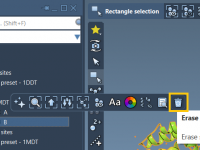
For molecular modelers, one of the key challenges is accurately modeling transitions between protein conformations. Whether you’re studying conformational analysis, working on free energy simulations, or preparing reaction coordinates, having a realistic and smooth transition path is essential. This is…
Mastering Node Selection in SAMSON with NSL.
Streamline Molecular Simulations with the Simulate Animation in SAMSON
For molecular modelers looking to conduct accurate and constrained simulations efficiently, the Simulate animation in SAMSON is an indispensable tool. Managing nanoscale movements and interactions often involves tedious setups that require synchronizing motions across various simulations. The Simulate animation provides…
Enhance Molecular Visuals with Ambient Occlusion in SAMSON.
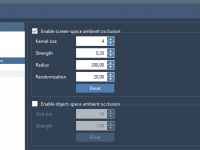
One of the challenges molecular modelers often face is accurately perceiving the depth of complex molecular structures. Distinguishing between overlapping or nested elements can be difficult, especially when working with highly detailed protein assemblies, ligand-receptor interactions, or molecular complexes. This…