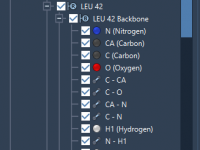
Unlocking Chain Attributes in SAMSON: A Practical Guide for Molecular Modelers
Effortlessly Exploring Binding Site Motions with Nonlinear Normal Modes in SAMSON
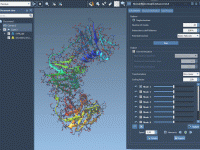
Molecular modelers often face the challenge of understanding large-scale structural motions, such as the opening of binding sites in biomolecules. This capability is crucial for elucidating functional mechanisms or exploring drug-target interactions. If you’re looking for an innovative and straightforward…
Discover How SAMSON Apps Simplify Molecular Modeling Tasks
Simplifying the Installation of SAMSON: A Step-by-Step Journey
For new users venturing into computational nanoscience, SAMSON provides a versatile platform perfect for life sciences, chemistry, physics, and more. However, getting started with a new software can sometimes feel overwhelming, especially with installation processes and system requirements. Here’s a…
Mastering Molecular Measurements in SAMSON.
Master the ‘Follow Atoms’ Animation in Molecular Modeling
Effortless Focus on Molecular Dynamics with ‘Look at Atoms’
Optimizing Molecular Geometries Faster with the FIRE Minimizer

For molecular modelers, achieving a stable, energy-minimized molecular geometry is a common goal before simulations or further structural analyses. However, traditional methods, such as the steepest descent algorithm, can be too slow when dealing with large-scale molecular movements. This inefficiency…