Simplifying Molecular Modeling: SAMSON’s Supported File Formats Explained

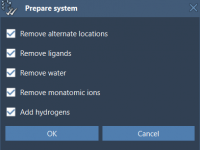
Molecular modeling often involves handling a variety of file formats, especially when dealing with complex datasets like molecular structures, trajectories, geometries, and more. For professionals and researchers in this field, managing and converting these formats can sometimes create unnecessary roadblocks.…