Efficient Molecular Pattern Design: Master Linear, Circular, and Curved Patterns in SAMSON

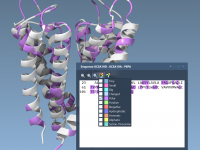
Designing molecular structures—especially in domains like nanotechnology or biomolecular modeling—can be a daunting task for anyone aiming for precision and efficiency. Whether you’re building nanotubes, modeling complex molecular arrays, or crafting intricate nanoscale patterns, SAMSON’s pattern editors offer a powerful…