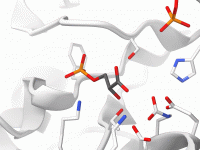
Streamline Molecular Manipulations with Move Editors in SAMSON
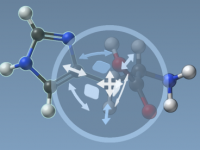
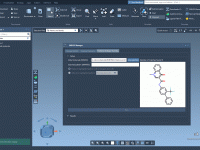
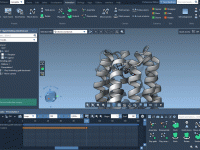
Molecular modeling often involves precise manipulations of structures: positioning, aligning, and transforming intricate molecular geometries can be a tedious task. SAMSON, the integrative molecular design platform, offers a variety of intuitive Move Editors that simplify this process for modelers. These…