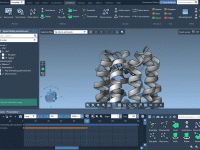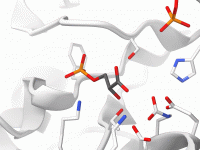
Decoding Property Model Attributes in SAMSON
Simplify Your Molecular Presentations with Background Animations
Effortless Perspective Changes: Managing Multiple Cameras in SAMSON
Streamlining Python Package Management in SAMSON
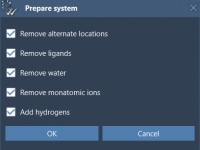
For computational molecular modelers, efficient scripting and workflow automation can feel cumbersome without a robust system to manage dependencies and libraries. Integrating specific Python packages into your modeling pipeline can open up incredible possibilities—from deep learning algorithms to advanced statistical…
A Practical Guide to Setting Up Umbrella Sampling Projects with GROMACS Wizard
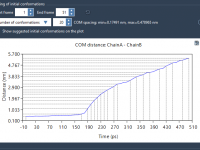
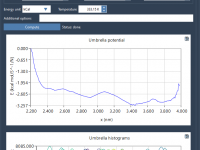
Umbrella sampling is a powerful technique in molecular modeling, enabling researchers to understand the energetics along a reaction coordinate. But implementing it can feel daunting, especially when you’re faced with setting up reaction-coordinate windows and managing simulations efficiently. With the…