Mastering Precise Molecular Manipulation with SAMSON’s Move Editors
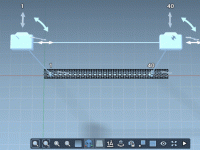
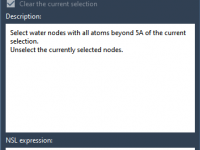
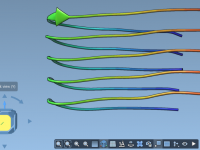
For molecular modelers, the ability to precisely manipulate molecular structures is often a crucial step in refining designs and visualizing interactions. However, achieving precise movements, rotations, and alignments can sometimes feel cumbersome or error-prone without the right tools. Fortunately, SAMSON’s…