Simplify Molecular Modeling with the Viewport Menus in SAMSON.

Pausing Presentations in Molecular Modeling Simplified
Visualizing Biological Assemblies: A Guide for Molecular Modelers
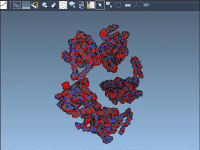
For molecular modelers, understanding how proteins organize themselves within their crystal structures or biological assemblies is essential. Whether it’s exploring interactions at protein-protein interfaces, reconstructing quaternary structures for simulation, or designing symmetric protein complexes, these insights are key to driving…