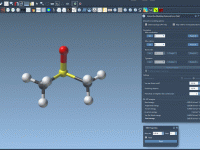
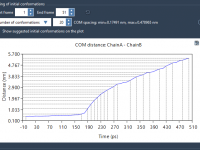
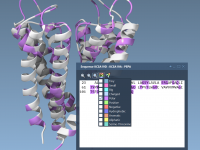
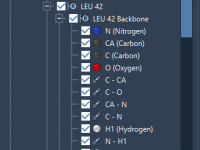
Highlighting Molecular Patterns for Efficient Analogue Design
For molecular modelers, the ability to strategically modify specific portions of a molecule can be critical for understanding and optimizing molecular properties. Whether it’s improving binding affinity, reducing toxicity, or analyzing structure-activity relationships, having precise control over pattern selection is…