Mastering Document Hierarchies in SAMSON: Tips for Molecular Modelers
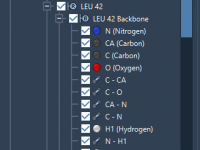
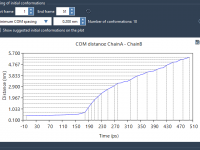
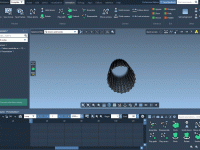
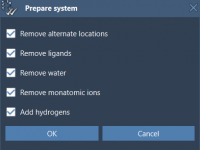
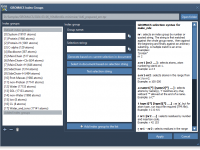
For molecular modelers, organization is key. When managing complex molecular data, having an intuitive and robust structure can significantly improve productivity. In SAMSON, the integrative molecular design platform, Documents provide a powerful hierarchical framework to store and manage your molecular…