Troubleshooting SAMSON Installation: Invalid Key or Credentials?
How to Define Replaceable Patterns in Molecules Using SMARTS Codes
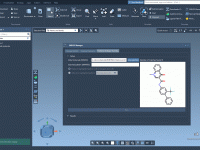
How Bond Order Filters Could Save You Hours in Molecular Editing
Designing Molecules in 2D While Staying in Sync with 3D
Avoid Common Pitfalls When Setting Up Production MD Simulations
One challenge that many molecular modelers face with molecular dynamics (MD) simulations is ensuring continuity and consistency between equilibration and production runs. It may sound trivial, but mistakes in setup parameters—particularly when switching from NPT or NVT equilibrations to production—can…
How to Save Time Designing Complex Polymer Sequences
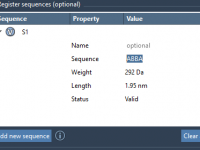
Saving Molecular Structures: What You Need to Know About SAMSON Exporters
Smooth Molecular Motion: A Practical Guide to Atom Animation in SAMSON
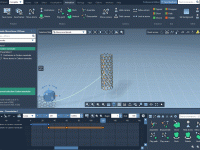
When preparing scientific presentations or molecular animations, one common frustration faced by structural biologists and molecular modelers is presenting complex atomic transformations in a clear, smooth, and repeatable way. Often, the motion of molecules is illustrated frame-by-frame, requiring tedious positional…