Refining Molecular Presentations with the Stop Animation in SAMSON
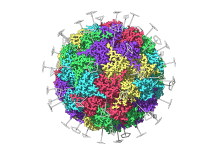
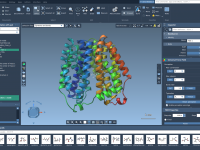
Molecular presentations play a crucial role in conveying complex structural insights, whether you are presenting your findings to colleagues, teaching students, or preparing professional reports. However, one common challenge that molecular modelers face is creating clear, segmented animations that allow…