Understanding Backbone Attributes in the Node Specification Language (NSL).
Streamlining Molecular Model Presentation Attributes in SAMSON
Clarifying Animation Attributes in SAMSON’s Node Specification Language
Simplify Molecular Analysis with Side Chain Attributes in NSL
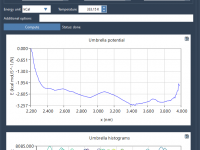
Mastering PMF Analysis with GROMACS Wizard
Effortlessly Generate Analogue Series Using the SMILES Manager.
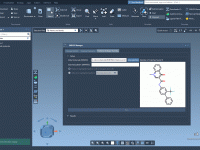
Integrate Python to Automate Molecular Modeling in SAMSON
Mastering Node Selection in Molecular Modeling.
For many molecular modelers, the precision of their work depends heavily on accurately selecting specific atoms, residues, molecules, or other structural entities. Whether you’re inspecting properties, measuring geometry, or editing molecular structures, mastering node selection is essential for effective workflows.…