





Visualizing Molecular Interactions with Contact Maps and Series
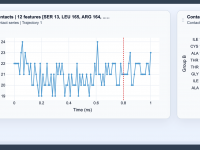
Molecular modelers often face challenges in tracking and understanding the subtle interactions between different regions of biomolecules over time. Identifying when key interface regions form or break, understanding domain interactions, or observing binding events are critical tasks in molecular dynamics…
Customizing Bond Graphs in Your Molecular Simulations
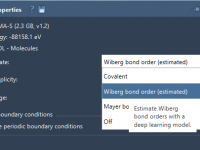
For molecular modelers, refining and understanding bond structures during simulations can be a challenging task. Whether you’re optimizing molecular geometries, exploring chemical reactivity, or examining material properties, the way bonds are updated can significantly affect your insights. This blog post…
Running Molecular Simulations in the Cloud with GROMACS Wizard
One common challenge for molecular modelers is ensuring they have enough computational resources to run molecular dynamics simulations. These simulations can demand significant computational power, especially when dealing with extensive systems, long timescales, or advanced workflows like umbrella sampling or…
Exploring Light Attributes in SAMSON’s NSL
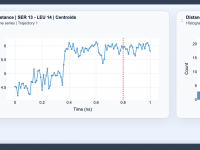
Understanding Molecular Interactions with Distance Analysis in SAMSON
Efficiently Structuring Molecular Presentations with the Stop Animation
Creating compelling presentations for molecular modeling can be challenging if you need to clearly communicate complex processes or highlight specific structural properties. One important pain point for molecular modelers is being able to pause and segment presentations effectively while maintaining…
Creating Smooth Protein Transition Paths with ARAP Interpolation
One of the recurring challenges for molecular modelers is analyzing conformational changes in proteins. Whether it’s modeling transition states for drug discovery, preparing reaction coordinates for free energy simulations, or simply visualizing conformational dynamics, obtaining a realistic, continuous transition pathway…
Streamline Molecular Simulations with Batch Mode in GROMACS Wizard
Molecular simulations often involve repetitive workflows, such as energy minimization, equilibration, and molecular dynamics across multiple conformations or systems. Configuring these setups manually for every iteration can be time-consuming – especially if dealing with dozens or hundreds of starting points!…