






Understanding Structural Model Attributes in SAMSON
Enhancing Molecular Models with Clear Labels and Annotations
In molecular modeling, clarity is crucial, especially when sharing findings or preparing visualizations for presentations. A recurring challenge for modelers is ensuring that annotations, measurements, or other textual elements remain readable and enhance the understanding of structures. SAMSON makes this…
Mastering System Neutralization for Coarse-Grained Simulations in GROMACS Wizard

Streamline Molecular Modeling with Camera Attributes in NSL
Managing complex molecular designs can be challenging, especially when dealing with intricate visualization setups. A well-tuned camera setup is crucial for controlling viewpoints and achieving the clarity needed to perform detailed analyses. SAMSON’s Node Specification Language (NSL) offers camera attributes…
Simplifying Molecular Model Selection with Node Group Attributes in SAMSON
Simplify Molecular Modeling with Backbone Attributes in SAMSON
Molecular modelers often face the mammoth task of sifting through complex molecular structures to find specific atom groups or perform focused simulations. SAMSON’s Node Specification Language (NSL) offers an elegant solution: backbone attributes. These attributes allow precise identification and manipulation…
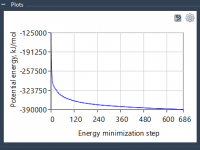
Streamline Molecular Modeling with Energy Minimization in SAMSON’s GROMACS Wizard
Streamline Your Molecular Models with Dock Animation in SAMSON
For molecular modelers, precise positioning of atoms and molecules into designated conformations is a frequent challenge. Whether simulating interactions or creating biologically accurate presentations, the importance of accurately docking groups of atoms or meshes cannot be overstated. This is where…
