Simplify Molecular Pathway Analysis by Exporting Selected Atomic Trajectories
How to Fix Small Icons on High DPI Monitors in SAMSON

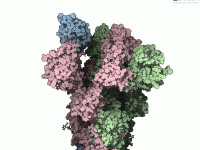
Visualizing the Motion of the SARS-CoV-2 Spike Protein
A Straightforward Guide to Installing SAMSON on Any Operating System
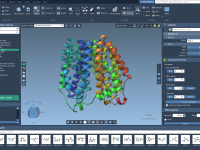
Step-by-Step Guide to Setting Up Universal Force Field (UFF) in SAMSON.
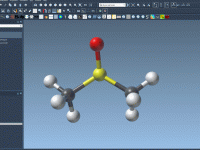
Mastering SAMSON AI Commands for Molecular Modeling Efficiency
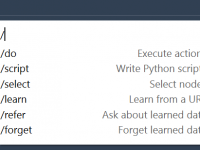
For molecular modelers, efficiency is crucial. Whether analyzing complex molecular structures, preparing simulations, or refining atomic interactions, streamlining tasks can save valuable time and effort. With SAMSON AI, a powerful assistant integrated within the SAMSON molecular design platform, users gain…
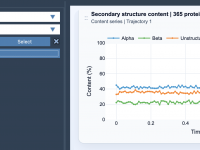
Enhancing Molecular Transition Paths with the Parallel Nudged Elastic Band Method
Streamlining DNA Nanostructure Simulations with Adenita Export Features
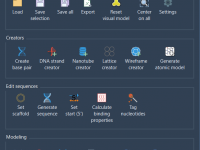
Designing DNA nanostructures is an intricate process, but equally important is preparing designs for simulation workflows. For molecular modelers working with coarse-grained simulation tools like oxDNA, exporting these nanostructures in a suitable format often presents a significant hurdle. With Adenita,…