





Boost Your Molecular Modeling Workflow with SAMSON Extensions

Understanding Residue Polarity for Molecular Modeling
Mastering Sequence Selection Across Views in SAMSON
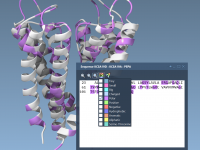
Molecular modelers often face the challenge of managing and selecting specific residues in complex molecular structures. Whether you’re focusing on a particular chain, analyzing biophysical properties, or preparing data for workflows, accurate and synchronized selection across multiple views is crucial.…
Mastering Carbon Nanotube Modeling with Precision

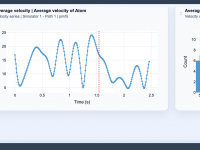
Exploring Molecular Dynamics with Average Velocity Insights
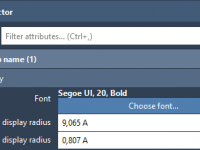
Mastering Label Customization for Clearer Molecular Models in SAMSON
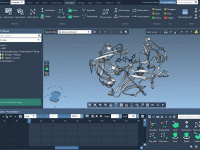
Navigating Your First Steps with SAMSON: A Beginner’s Guide
Effortless Node Visibility Control with Flash Animation in SAMSON
For molecular modelers, managing the visibility of molecular nodes during animations can frequently be a cumbersome task, especially when trying to visually emphasize or simplify complex molecular interactions. Thankfully, SAMSON’s Flash animation feature offers an effective and straightforward solution tailored…