





Efficiently Managing Hidden and Visible Nodes in SAMSON
Streamline Molecular Visualization with Custom Visual Presets
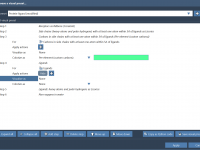
Mastering Conformation Attributes in SAMSON for Precise Molecular Modeling
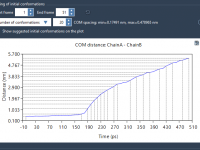
Efficient Geometry Refinement with UMA Force Field in SAMSON
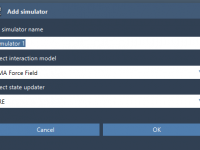
For molecular modelers, achieving accurate geometry refinement quickly can be a challenging task. Whether you’re exploring molecular structures, refining inorganic materials, or simulating catalyst systems, the UMA Force Field in SAMSON provides a machine-learning-based solution designed for high responsiveness and…
Practical Steps for Molecular Building with Atoms in SAMSON
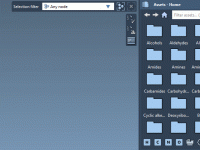
Leveraging Path Attributes in SAMSON’s Node Specification Language
Simplifying Umbrella Sampling with GROMACS Wizard for Molecular Modeling
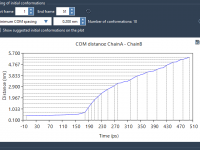
For molecular modelers, conducting detailed simulations to analyze reaction pathways can be an intricate and time-intensive task. One specific challenge emerges when converting a reaction-coordinate pathway into meaningful sampling windows for Potential of Mean Force (PMF) analysis. The GROMACS Wizard…
Streamline Protein Analysis with Ramachandran Plotting in SAMSON
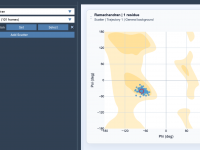
Understanding protein backbone conformations is essential for molecular modelers aiming to study secondary structures, conformational preferences, or identify anomalies. The Ramachandran plot is a tried-and-tested tool for this task, but integrating it effectively into your analysis pipeline can sometimes be…
Say Goodbye to Noisy Data: Smoothing Insights with Density Curves
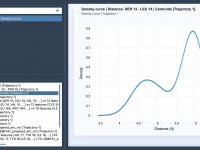
Molecular modeling often involves working with noisy datasets—scattered, uneven distributions that can make it challenging to extract meaningful trends. Whether you’re analyzing energy variations, distances, or structural properties, addressing such noise is critical to making sense of molecular landscapes. This…