





Simplify Molecular Zooming With the Zoom Camera Animation in SAMSON
Boost Your Molecular Insights with Path Analyzer in SAMSON.
One of the significant challenges molecular modelers face is analyzing the complexity of trajectories or paths for deeper insights—especially when it comes to understanding molecular conformations and interactions over time. Luckily, SAMSON, the integrative molecular design platform, offers a robust…
Streamlining Path Exports for Molecular Simulations
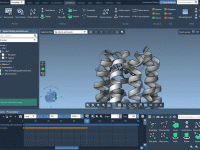
Build Precise Molecular Patterns with Intuitive Tools in SAMSON
Building large molecular systems efficiently and accurately can sometimes feel like solving a puzzle with too many pieces. Whether designing nanotubes for nanotechnology, biomolecular assemblies for biological simulations, or modeling complex materials for material science, creating repetitive structures is an…
Essential Tips for Configuring Molecular Dynamics Parameters in GROMACS Wizard
Demystifying File Attributes in SAMSON’s Node Specification Language
Streamline DNA Nanostructure Prototyping: Importing Designs into Adenita
DNA nanostructures offer immense opportunities for structural and functional innovation in nanotechnology. However, starting every nanostructure from scratch can be overwhelming and time-consuming for researchers and modelers. Luckily, Adenita, an extension for SAMSON, makes this process far more efficient by…
Mastering Molecular Dynamics with the ‘Look at Atoms’ Animation
Effortlessly Separate Atoms with UNDOG for Molecular Modeling
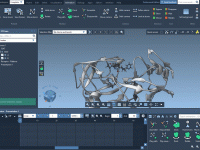
For molecular modelers, efficiently separating groups of atoms, meshes, or structural nodes can often present a significant challenge. Whether you are working on ligand-receptor interactions, generating molecular presentations, or simulating animations, achieving clarity in visualization is paramount. The Undock animation…