Author: OneAngstrom
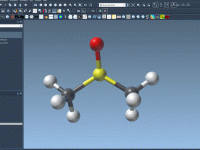
Effortlessly Running GROMACS Simulations in the Cloud with SAMSON’s GROMACS Wizard
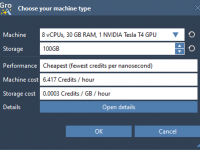
Molecular modelers often face a recurring challenge: running heavy computations on local machines. These simulations, particularly involving molecular dynamics, may require significant computational resources and time. SAMSON’s GROMACS Wizard provides a practical solution by allowing users to perform these demanding…
Streamlining Molecular Modeling with SAMSON Node Types
Mastering Residue Secondary Structure Queries in SAMSON’s NSL
Streamlining Molecular Trajectories with Reverse Path Animation in SAMSON
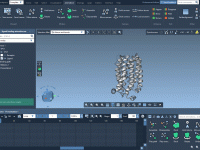
When studying molecular dynamics, visualizing trajectories and conformational pathways is essential. Whether you’re analyzing how a biomolecule shifts between states or testing novel computational experiments, efficiently reviewing your data is key. One challenge molecular modelers often face is replaying trajectories…
Mastering Animation Attributes in SAMSON’s NSL
Molecular modeling often involves navigating large and complex molecular systems, especially when working with animations to visualize processes. If you’re using the SAMSON molecular design platform, understanding Animation attributes in the Node Specification Language (NSL) can simplify this process significantly…
Streamline Your Computational Workflows with SAMSON Job Manager
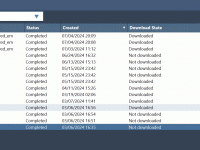
For molecular modelers, running complex computations in the cloud can be a game-changer. However, managing these computations—tracking progress, accessing results, and keeping everything organized—can quickly become overwhelming. The SAMSON Job Manager provides a clean and efficient way to address this…