Author: OneAngstrom
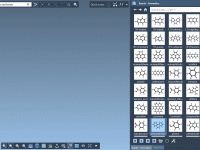
Leveraging Molecular Fragments to Build Complex Structures Efficiently
Ensuring Compatibility of SAMSON Extensions with Versioning Policies
An Academic Advantage with SAMSON.
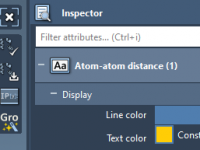
Unveiling the Power of Structural Model Attributes in Molecular Design
Mastering Constrained Simulations in SAMSON’s Simulate Animation
For molecular modelers, achieving accurate simulations that adhere to constraints can often feel like threading a needle. Have you ever encountered challenges ensuring your simulations respect specific atomic positions or move seamlessly under constraints? SAMSON’s Simulate animation offers an effective…
Simplifying Molecular Design with Editor Tools in SAMSON

Simplified Setup of the Universal Force Field in SAMSON
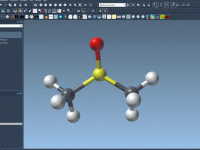
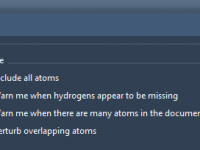
Molecular modelers often encounter challenges when preparing simulations involving complex systems. These challenges include automatically perceiving molecular structures, computing bonds, atom types, and more. If you’re using SAMSON for molecular modeling, the Universal Force Field (UFF) offers a robust solution…