





Author: OneAngstrom
Effortlessly Designing Molecular Patterns with SAMSON’s Pattern Editors
Creating complex molecular structures can often feel overwhelming for molecular modelers. Repetitive tasks like arranging molecules in precise patterns to form nanotubes, circular chains, or other nanoscale architectures consume time and effort. SAMSON addresses this challenge with its intuitive Pattern…
Streamline Your Simulations with the Simulate Animation in SAMSON
Mastering Molecular Visualizations with Custom Visual Presets in SAMSON

For molecular modelers and researchers, achieving the ideal visualization of complex molecular systems can often feel daunting. Simplifying these intricate representations without losing clarity or precision is a common pain point. SAMSON, the integrative molecular design platform, tackles this effectively…
Effortless Bond Manipulation with IM-UFF in SAMSON
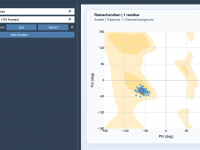
A Practical Guide to Ramachandran Plots for Protein Modeling
Understanding Path Attributes in SAMSON’s Node Specification Language
Molecular modeling often involves handling complex data to accurately represent molecular structures and properties. When working with large datasets, ensuring you can efficiently specify and query features is essential. SAMSON’s Node Specification Language (NSL) offers a powerful way to streamline…
Mastering the Pause Animation for Seamless Molecular Presentations
Effortlessly Manage Molecular Models with SAMSON’s Document View
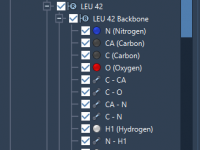
Mastering Custom Index Groups in GROMACS Wizard: Simplify Complex Molecular Workflows
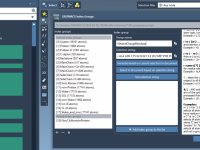
Molecular modelers often encounter scenarios where standard index groups automatically generated by GROMACS just aren’t enough. Whether you need to isolate specific residues, define pull groups, or prepare for advanced analysis, creating custom index groups can significantly streamline your workflow.…