Author: OneAngstrom
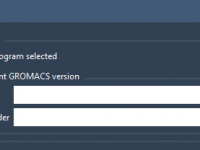
Streamlining Molecular Simulations with Custom GROMACS Versions
Step-by-Step Guide to Interactive Nanotube Modeling with SAMSON

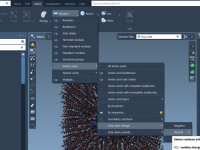
Streamlining Batch Computations with GROMACS Wizard
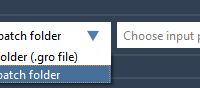
Performing molecular simulations efficiently becomes crucial when working with multiple related systems or conformations. Instead of setting up workflows manually for each project, imagine leveraging automation to batch process computations seamlessly. Enter the GROMACS Wizard Batch Computations in the SAMSON…