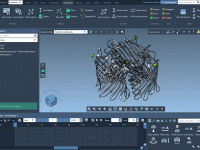
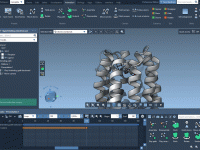
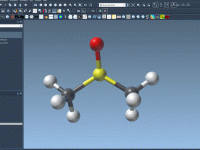
Efficiently Save and Reuse Your DNA Designs in Adenita
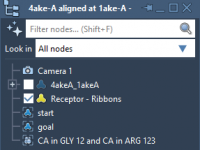
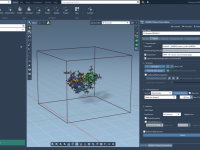
One common challenge faced by molecular modelers is managing complex DNA structures across multiple projects. Often, meticulously designed components are either reconstructed from scratch or inconsistently saved, wasting valuable time and effort. With Adenita, SAMSON’s integrative tool for designing DNA…