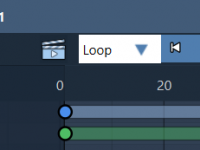
Making Molecular Models Gradually Disappear: A Guide to the Disappear Animation
As a molecular modeler, presenting your work effectively is critical, whether for research, education, or communication with peers. Clear animations can make complex molecular structures and changes comprehensible. One common challenge is progressively fading structures out during presentations or instructional…
Mastering Undo and Redo in Molecular Modeling
Simplify Your Molecular Simulations with Custom Index Groups
Exploring Chain Attributes in SAMSON for Molecular Modeling Efficiency
Demystifying Conformation Attributes in Molecular Modeling
A Step-by-Step Guide to Building Carbon Nanotube Models in SAMSON

Effortlessly Track Molecular Movements with ‘Follow Atoms’ Animation in SAMSON
Enhancing Molecular Transparency with Appear Animations in SAMSON
For molecular modelers working on creating intuitive and visually appealing molecular designs, effectively managing transparency can be a significant challenge. Imagine you're presenting a complex molecular structure with layered details. You want certain nodes to progressively appear, emphasizing specific parts…