





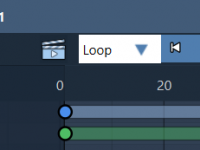
Easily Create Molecular Rocking Animations
For molecular modelers, creating visually engaging presentations or simulations is often essential for communicating molecular dynamics, insights, or structural properties. But how do you animate movement effectively, while ensuring precision and clarity? SAMSON’s Rock animation offers a clean and accessible…
Efficient Camera Node Selection in SAMSON with NSL
Understanding the SARS-CoV-2 Spike’s Critical Opening Motion
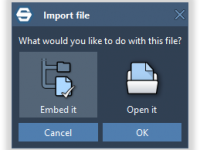
Embedding Files and Folders in SAMSON Documents: A Game-Changer for Molecular Modelers
Decoding Segment Attributes in SAMSON for Molecular Modeling
Defining the Sampling Box for Ligand Unbinding Pathways Made Easy
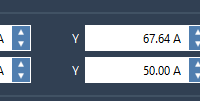
When working on molecular simulations, accurately capturing ligand unbinding pathways can be challenging, especially when defining the region for sampling. A well-defined sampling region ensures efficient exploration of the ligand’s motion and reduces computational overhead. In this post, we’ll take…
Simplify Your Molecular Animations with the Show Effect.
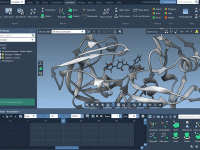
How to Interactively Simulate Molecules in SAMSON.
Streamline Vertical Camera Movements with the Pedestal Camera Animation.
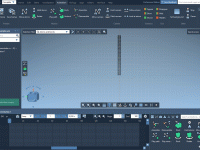
Molecular modeling often demands precise control over the viewpoint, especially when exploring structures closely or presenting data effectively. Adjusting a camera's position within a three-dimensional molecular scene is time-intensive and can interrupt the modeling workflow. This is where the Pedestal…