How to Share and Manage Molecular Design Documents on SAMSON Connect
Collaborative work in molecular design often involves sharing documents like models, simulations, or research reports with colleagues, research groups, or external partners. SAMSON Connect offers robust features for securely sharing and managing documents, streamlining collaboration across diverse projects—all while giving…
Progressively Revealing Atoms in Molecular Animations
Efficiently Pause Animations in Molecular Presentations
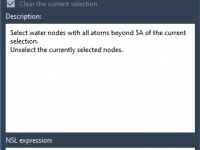
Choosing Which Crystal Waters to Keep in Molecular Simulations with GROMACS Wizard.
Streamlining Molecular Modeling with SAMSON’s Editors

Mastering Custom Index Groups in GROMACS Wizard
Faster Molecular Geometry Optimization with the FIRE Minimizer

Optimizing molecular geometry is a critical, time-intensive task for molecular modelers, enabling stable and realistic molecular structures that are essential for simulations and structural studies. Traditional methods like the steepest descent algorithm can sometimes fall short, particularly when dealing with…