






Simplify Your Molecular Modeling with Light Attributes in SAMSON
Understanding Property Model Attributes in SAMSON’s NSL
If you're a molecular modeler, you've probably encountered challenges in efficiently organizing, identifying, and filtering nodes or components within complex molecular models. SAMSON's Node Specification Language (NSL) provides a solution to this with its property model attribute space, abbreviated as…
Simplify Your Workflow with SAMSON Extensions

Spotting Molecular Stability with Contact Persistence Analysis
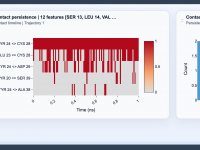
For molecular modelers, identifying stable and fleeting interactions within molecular trajectories is often key to understanding biological mechanisms or refining molecular designs. But how can one systematically uncover which interactions are persistent and which are just transitory? Enter the Contact…
Effortless Camera Switching in Molecular Models
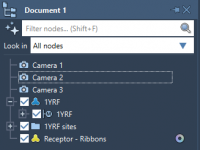
Easily Synchronize Molecular Paths with the Play Path Animation in SAMSON
For molecular modelers dealing with intricate trajectories, structures, or conformational changes, reviewing molecular animations smoothly can be challenging. The Play Path animation in SAMSON is an efficient feature that simplifies the visualization of molecular paths, cycles, and transitions within a…
Understanding SARS-CoV-2 Spike Motion: Simulating Transitions with SAMSON
Enhancing Molecular Modeling: Exploring Defects in Diamond Structures
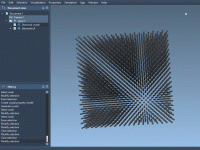
Molecular modelers working with crystal structures often need to simulate imperfections within those structures to better understand their real-world behavior. For instance, defects in a diamond crystal can significantly influence its physical properties. The Crystal Creator Extension in SAMSON provides…
Unveiling Correlations with Custom Scatter Plots in SAMSON
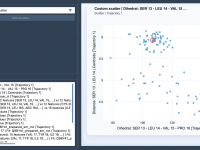
Molecular modelers often face the challenge of analyzing large datasets quickly and effectively to uncover meaningful relationships between properties. Identifying correlations, anticorrelations, or clusters can lead to groundbreaking insights, but the process isn’t always straightforward. If you’ve struggled with making…