





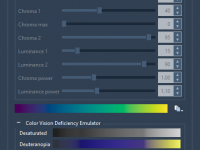
Navigating Molecular Shapes with Atom Geometry in SAMSON.
Effortlessly Manage Collaborative Groups in SAMSON Connect
Bring Molecular Models to Life with the Rock Animation in SAMSON
Master the Subtle Art of Molecular Visibility: The ‘Disappear’ Animation in SAMSON
Demystifying Path Attributes in SAMSON’s Node Specification Language
A Seamless Start with SAMSON: Simplify Your Setup Process.
Streamlining Molecular Modeling with Backbone Attributes in SAMSON
Molecular modeling often involves navigating complex datasets, identifying specific molecular components, and interpreting their traits. For researchers and designers working with molecular structures, efficiently filtering and querying data is crucial for productivity. The backbone attribute space in the Node Specification…
Making Energy Analysis Easier: Leveraging Views in Path Analyzer
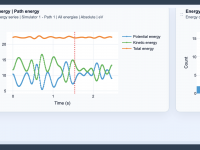
Molecular modelers often face challenges when trying to interpret energy data to better understand molecular pathways and interactions. Comparing relative energy differences or identifying trends can become a tedious task, especially when balancing various energy components, units, and visualization preferences.…