Effortlessly Manage Molecular Simulations in the Cloud
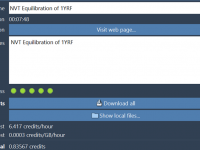
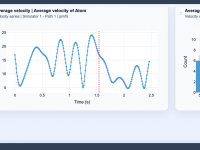
For molecular modelers, managing complex simulations—whether it’s protein structure prediction or molecular dynamics—is a critical aspect of their workflow. However, dealing with large datasets, ensuring cloud resources are properly utilized, and retrieving results efficiently can turn into a cumbersome process.…