Author: OneAngstrom
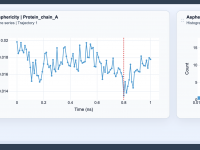
Visualizing Molecular Shape Changes with Asphericity Analysis
Streamlining Python Package Management in SAMSON.
For molecular modelers and scientists leveraging Python scripting in SAMSON, managing Python packages efficiently can be a game-changer. Whether you’re integrating specialized libraries for deep learning, bioinformatics, or advanced simulations, SAMSON’s built-in Python Package Manager simplifies the process, ensuring that…
Simplifying Molecular Modeling with SAMSON Extensions

Effortless Residue Analysis with SAMSON’s NSL
Molecular modeling presents challenges, especially when you need to select or filter specific residues based on their attributes. Whether you are examining large biomolecules or fine-tuning designs for computational experiments, manually processing residues is time-consuming. Fortunately, SAMSON’s Node Specification Language…
Mastering the Zoom Camera Animation in Molecular Modeling
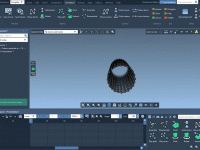
Molecular modeling requires precise control of visual perspectives, particularly when presenting results or fine-tuning molecular structures. A common challenge? Achieving seamless zoom effects while maintaining the target point of focus. For those using the SAMSON molecular design platform, the Zoom…
Streamlining Protein Cleaning with Batch Protein Preparation
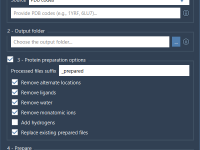
For molecular modelers working with multiple PDB files, the challenge of preparing protein structures efficiently can be daunting. Missing residues, alternate locations, or unnecessary molecules often require extensive manual work before docking, simulation, or other workflows can begin. Thankfully, SAMSON…
Dealing with Network Proxy Settings During SAMSON Installation.
Understanding Structural Changes with RMSD Analysis in SAMSON
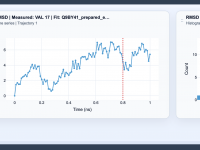
One frequent challenge in molecular modeling is distinguishing between overall rigid-body motion and meaningful internal structural changes. This distinction is crucial for making sense of conformational dynamics, binding events, and other molecular processes. The RMSD (Root Mean Square Deviation) analysis…