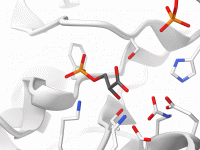
Streamlining Polymer Design with Sequence Registration
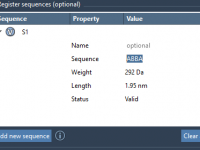
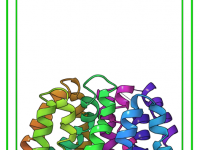
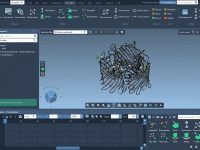
One of the challenges in molecular modeling and materials design is creating complex, custom polymers with repeating sequence patterns. Whether you’re constructing synthetic polymers for material property predictions or drug-polymer conjugates, manually assembling these structures can be tedious and error-prone.…