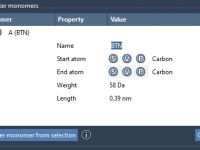
Save Time and Enhance Visualizations with SAMSON’s Visual Presets
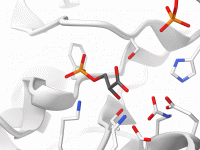
Introduction to Visual Presets For molecular modelers, visualizing complex molecular systems effectively and efficiently is key. However, applying detailed visual representations and color schemes can quickly become painstaking, especially when dealing with intricate molecular structures. This is where SAMSON’s Visual…