Exploring Relationships with Custom Scatter in Molecular Design
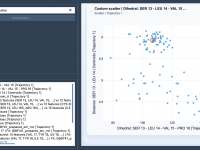
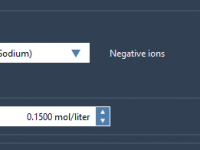
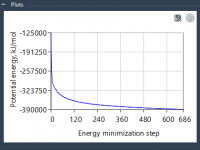
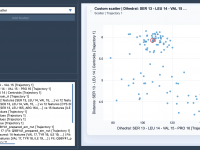
For molecular modelers, analyzing relationships between variables can be a critical part of gaining scientific insights about molecular systems. Whether you’re investigating correlations between energy and distance or uncovering clusters in complex data, visualizing these connections is essential. With SAMSON’s…