Author: OneAngstrom
Practical Strategies for Colorizing Molecular Models in SAMSON

Mastering Pause Animations for Better Molecular Presentations
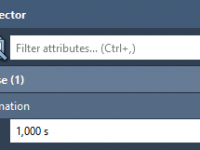
Master Transparency Effects with the Pulse Animation
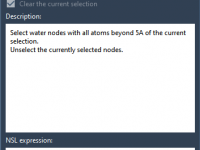
Mastering the Node Specification Language for Advanced Molecular Modeling
Exploring Residue Polarity in Molecular Modeling
In molecular modeling, understanding the polarity of residues is essential for comprehending their biochemical functions and interactions. Residue polarity determines how a molecule interacts with its environment, influencing functions like solubility, binding affinity, and overall molecular stability. Let’s explore how…