Author: OneAngstrom
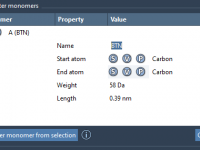
Importing Molecules into SAMSON: A Practical Guide for New Users

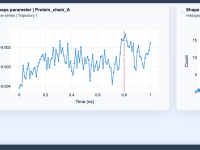
Understanding Molecular Shape Trends with the Shape Parameter
Boost Your Molecular Modeling Skills with SAMSON’s Interactive Tutorials
When starting with molecular modeling, mastering complex software interfaces can be daunting. SAMSON, an integrative molecular design platform, offers a unique feature to address this challenge: interactive tutorials. These tutorials provide hands-on guidance within the application itself, making it easier…