Author: OneAngstrom
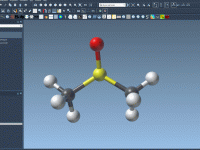
Mastering Universal Force Field (UFF) Setup in SAMSON
Building Custom Lipid Layers Around Proteins Using Molecular Box Builder
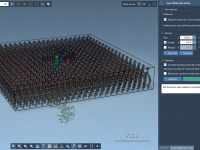
Streamline Molecular Pathways with the Parallel Nudged Elastic Band Method
For molecular modelers, identifying accurate transition paths in complex systems is often a critical task. Whether you’re studying protein-ligand interactions or conformational changes in macromolecules, refining initial guesses into meaningful pathways can be a daunting challenge. The Parallel Nudged Elastic…
Building Reusable Polymer Sequences in SAMSON
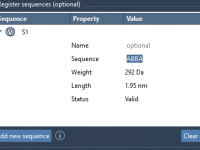
Simplify Structural Adjustments with the Undock Animation
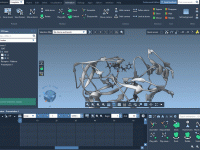
Elevate Your Molecular Visuals with the Pedestal Camera Animation
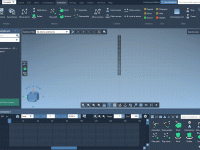
Streamline Molecular Modeling with Quick Groups in SAMSON
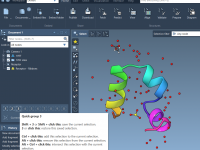
Understanding Molecule Attributes in SAMSON’s Node Specification Language (NSL).
Molecular modeling often requires precise filtering and organization of molecules for tasks such as simulation, visualization, and property analysis. If you’ve ever wondered how to efficiently categorize or interact with molecules based on specific characteristics, SAMSON’s Node Specification Language (NSL)…